Chapter 1
Relativistic Effective Potential Development
1
Introduction
Poor excitation energies for uranium V, U4+,
and other actinide cations as well as an anomalous bond length
for the uranyl cation, UO2 2+,
reported at the Second Conference of the
Department of Energy Computational Grand Challenge Application:
Relativistic Quantum Chemistry of Actinides [],
motivated an investigation of uranium
relativistic effective potentials (REPs).
The tradeoff between the incorporation of relativistic effects,
which stem from the inclusion of an atomic subshell in the REP,
and both
the accommodation of orbital polarization and the explicit treatment
of electron correlation,
which are permitted by the inclusion of an atomic subshell in the
valence space,
is examined at the self-consistent field (SCF)
and the configuration interaction (CI), both with and without
spin-orbit (SO) coupling,
levels of theory using
several new U REPs, with decreasing core sizes,
developed by P. A. Christiansen.
2
Results and Discussion
2.1
Uranyl Cation
Chapter presents background material on UO2 2+.
Table shows uranyl bond lengths using the five uranium REPs.
For the 78 and 68 REPs the basis set is converged.
For the 62 and 60 REPs the calculations are not at the basis set limit.
As is well known the SO interaction does not contribute to the bond length,
as comparison of the SCF and SOCIS results indicates.
de Jong bond lengths are from triple zeta quality basis sets.
No doubt these are not at the basis set limit.
The C68 REP gives the best bond length in comparison.
The spread between the REPs values is approximately 0.03 Å.
Table shows uranyl bond lengths using the five uranium REPs
with the (12s 11p 10d 8f 1g)/[12s 11p 10d 8f 1g]
basis set from the Stuttgart group [].
Previous claims concerning bond lengths converged with respect to
the basis set may have been premature.
| REP | SCF | SOCIS Active Space |
| | 6s 6p 5f | 5d 6s 6p 5f | 5p 5d 6s 6p 5f |
| E78 | 3.084 | 3.084 | | |
| C78 | 3.091 | 3.091 | | |
| C68 | 3.125 | 3.125 | 3.127 | |
| C62 | 3.142 | 3.144 | 3.114 | 3.110 |
| C60 | 3.141 | 3.144 | 3.110 | 3.106 |
| DHF [] | 3.120 |
| CCSD(T) [] | 3.241 |
Table 1:
UO2 2+
Bond Lengths in Bohr
| REP | SCF | SOCIS Active Space |
| | 6s 6p 5f | 5d 6s 6p 5f | 5p 5d 6s 6p 5f |
| E78 | 3.076 | 2.917 | | |
| C78 | 3.083 | 2.921 | | |
| C68 | 3.120 | 3.043 | 3.044 | |
| C62 | 3.129 | 3.039 | 3.040 | 3.028 |
| C60 | 3.129 | 3.048 | 3.048 | 3.035 |
| DHF [] | 3.120 |
| CCSD(T) [] | 3.241 |
Table 2:
UO2 2+
Bond Lengths in Bohr With the Stuttgart
(12s 11p 10d 8f 1g)/[12s 11p 10d 8f 1g]
Basis Set
3
Conclusions and Future Work
The 5d10 subshell of U is best included in the valence space.
In lieu of larger basis sets, better treatment of electron correlation,
and more accurate DF uranyl bond lengths, the present results support
this conclusion.
U4+ excitation energies are unlikely to shift radically with an
improved ab initio treatment.
It is reasonable to expect that excitation energies using other core sizes will
shift comparably; the 68 REP energy levels will remain sandwiched between
those of the 78 and 62 REPs, and will probably remain in best agreement with
experiment.
Furthermore, substantial other REP and pseudopotential work indicates
that for most accurate results the 5d10 subshell should be in
the valence space [,,,,].
The uranyl results are not so comforting.
A straightforward path to further study of the uranyl bond lengths
covers three points:
(1)
SCF basis set convergence, that is, for both DF and HF/AREP;
this should be easily attainable using numerical SCF programs;
systematic basis set improvement for actinides is problematic,
Chapter , and overdue; hopefully, this can be remedied.
(2)
A systematic analysis of core-valence correlation to elucidate
the role, if any, of the 5d10, the 5p6, and the
5s2 subshells in the uranyl bond length, as in [];
of course, this requires basis sets;
otherwise however, because uranyl is a closed shell system,
the present programs, in particular SOGUGA, should be adequate.
(3)
Four- and two-component calculations with and without frozen core
orbitals to differentiate relativistic and frozen core effects
and to identify core-valence partitioning problems, if any;
the concern here is to pinpoint which REP approximations dominate
as was done in some of the early REP and shape-consistent
work [,].
Other actinyls should be studied similarly;
curium seems a logical choice because it is clearly more
like a lanthanide than an early actinide.
The 5p6 subshell of U is probably best included in the REP.
Here the tradeoff between incorporation of relativistic effects,
which stems from being in the REP,
and inclusion in the valence space, which allows for polarization and
treatment of correlation effects,
falls on the side of relativity.
The SO splitting of the 5p6 subshell is substantial,
yet it has little effect on U4+ excitation energies.
Removal of the 5p6 electrons from the REP, however,
greatly shifts all the energy levels.
The role of the 5p6 subshell in uranyl is unlikely to be significant.
1
Background
The early actinides exist in a wide range of oxidation states,
from II to VII [,].
The most stable oxidation states are:
III for actinium,
IV for thorium,
V for protactinium,
VI for uranium,
V for neptunium,
IV for plutonium,
and
III for the rest of the series with the possible exception of II for nobelium.
In aqueous solutions without complexing agents the penta- and hexavalent
cations are too acidic to exist as hydrated ions.
They hydrolyze to form weakly acidic dioxoactinide(V)
and moderately acidic actinyl cations, respectively.
The dioxoactinide(V) cations are known from Pa through Am.
The actinyl cations are known from U through Am.
The electronic absorption spectrum of AmO2 2+
has been observed in several acidic aqueous solutions:
nitric acid [],
perchloric acid [,,,],
sulfuric acid [,],
and other aqueous media [].
These spectra, as well as those of all other AnO2 2+ and AnO2 + cations,
contain two characteristic categories of transitions:
narrow, usually weak peaks
and
broad, usually strong peaks.
The narrow peaks are the actinide fingerprint
f ¬ f transitions;
the broad and strong peaks are ligand-to-metal charge transfer
excitations [].
The transitions are either gerade to gerade or ungerade to ungerade
for AnO2 2+ and AnO2 +, in general.
They are thus electric dipole forbidden and are vibronic, magnetic dipole,
or electric quadrupole in origin.
The selection rules for the latter are
DW = 0, ±1
and
DW = 0, ±1, ±2,
respectively.
For AmO2 2+ the different acidic aqueous media do not significantly alter
the transitions.
The following peaks have been observed:
10100 cm-1, strong and narrow;
15080 cm-1, narrow;
13200, 13660, 16160, 18250 cm-1, weak and broad;
22310, 22790, 23350, 24480 cm-1, a strong and broad band.
In not strongly complexing, acidic aqueous solution
AmO2 2+ is vibrant yellow; Am3+ is pink.
The absorption spectra of AmO2 2+ complexed with carbonate
is significantly different from that of the acidic environments;
the complex is red-brown [].
However, the complex with phosphate yields a spectrum that is similar
to those in acid [].
The electronic absorption spectrum of AmO2 +
has been observed in several aqueous solutions:
potassium carbonate, nitric acid, and sulfuric acid [],
perchloric acid [],
and other aqueous media [].
For AmO2 + the different aqueous media do not significantly alter
the transitions.
The following peaks have been observed:
13980, 19490 cm-1, strong and narrow;
24080, 27170, 28900 cm-1, weak and broad;
30600, 32100 cm-1, a broad band.
No assignments have been offered;
even speculation is sparse [,].
Searches for oxidation states higher than III for Cm have been
unsuccessful [].
2
Results and Discussion
The common uranyl closed shell core of AmO2 2+, AmO2 +, and CmO2 2+
is
1su2 1sg2 1pu4 2su2 2sg2 3su2 2pu4 1pg4 3sg2.
This is consistent with our group's other work.
The ordering is that of ascending orbital energy.
Various arguments from empirical data and ab initio studies
have established that 3su is the orbital from which the low-lying
excitations occur for UO2 2+.
Orbital energies are not quantum mechanical observables;
electron excitations are quantum mechanical observables.
In the SOCI calculations on uranyl, the lowest transition
is, in fact, from 3su [,].
The population analyses for AmO2 2+, AmO2 +, and CmO2 2+
are displayed in
Table .
Substantial 6p holes are found:
0.7, 0.5, and 0.3 electrons, respectively.
In AmO2 2+ and CmO2 2+ the metals have a partial charge of
+1.66 and +1.87, respectively, significantly lower than in
UO2 2+ [] and NpO2 2+ [],
so that the oxygens are slightly positive: +0.16 and +0.06.
In AmO2 + the charges are +1.29 for Am and -0.14 for O.
| Atom | s | p | d | f | g | Total |
| AmO2 2+ |
| Am | 1.88 | 5.32 | 1.70 | 6.42 | 0.005 | 15.33 |
| O+O | 3.75 | 7.82 | 0.095 | - | - | 11.67 |
| AmO2 + |
| Am | 1.90 | 5.45 | 1.61 | 6.74 | 0.004 | 15.71 |
| O+O | 3.77 | 8.45 | 0.071 | - | - | 12.29 |
| CmO2 2+ |
| Cm | 2.08 | 5.68 | 1.47 | 6.89 | 0.003 | 16.13 |
| O+O | 3.74 | 8.06 | 0.070 | - | - | 11.87 |
| CmO2 + |
| Cm | 2.09 | 5.66 | 1.43 | 7.38 | 0.003 | 16.57 |
| O+O | 3.73 | 8.65 | 0.058 | - | - | 12.43 |
Table 1:
SCF/(cc-pVDZ Atom) Population Analyses for Lowest Calculable States
2.1
AmO2 2+
The MRSOCIS/cc-pVDZ electronic spectrum at the ground state MRSOCISD/cc-pVDZ
equilibrium bond length is listed in Figure .
du2fu1 4F3/2u is the ground state.
This state should have a negligible magnetic moment due to cancellation
of the spin and orbital contributions.
The 4Fu multiplet is regular and
has no interpenetrating states.
However, the SO splittings are in the ratio 1:4:4, not 1:1:1 as
expected [], suggesting some intermingling of states.
The rest of the spectrum is complicated.
In particular, the du1fu2 4Du multiplet is convoluted.
The first charge transfer states are the six 3su1 du2fu2 6Su
states.
This multiplet is inverted, and
its components are not evenly spaced.
The parenthesized values on the left are assignments of the observed
AmO2 2+ transitions.
The ungerade to gerade transitions begin at 36,000 cm-1 based on
MRSOCIS/cc-pVDZ calculations.
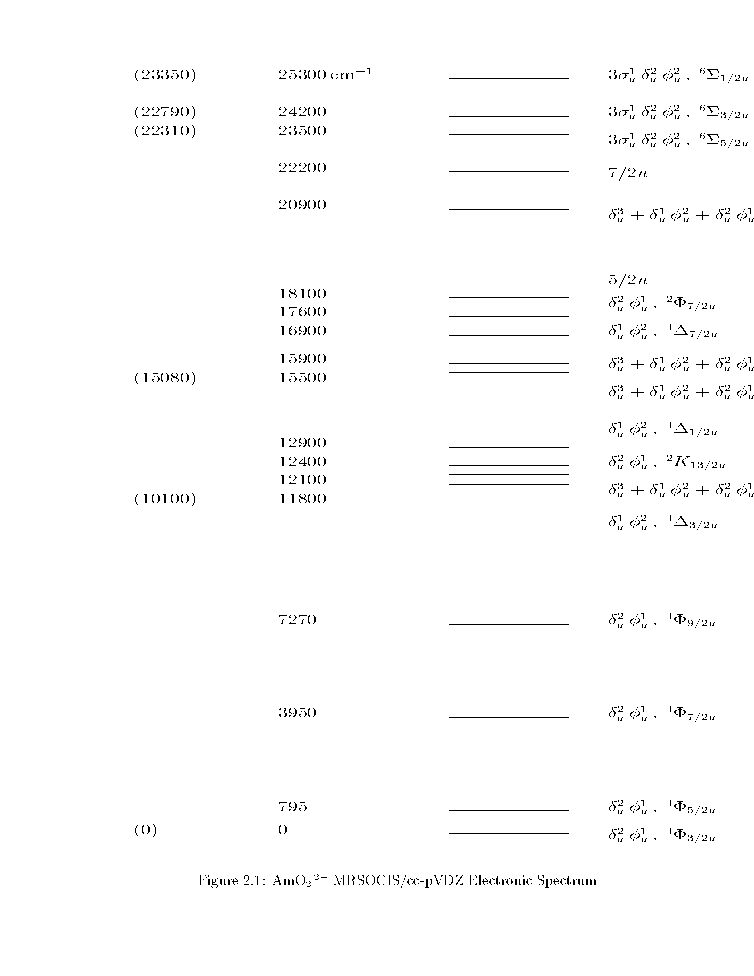
Figure 1:
AmO2 2+ MRSOCIS/cc-pVDZ Electronic Spectrum
At the SCF/cc-pVDZ level the du2fu1 4Fu
equilibrium bond distance, Re, is 1.53 Å.
Re for du1fu2 4Du is 1.52 Å.
At the MRSOCISD/cc-pVDZ level the du2fu1 4Fu
Re is 1.57 Å.
A symmetric stretch frequency of 1240 cm-1 is obtained from
a quadratic fit, Table .
The 56% relative error is large.
The most important sources of error are:
basis set quality, electron correlation treatment,
fitting method, and comparison to empirical data from solution.
The experimental frequency from crystalline sodium americyl acetate
is 749 cm-1 [].
2.2
AmO2 +
and
CmO2 2+
SCF calculations yield du2fu2 5S+g ground states.
The MRSOCIS/cc-pVDZ electronic spectra at the MRSOCISD/cc-pVDZ ground state
equilibrium bond lengths are listed in Figures
and .
du2fu2 5S+0+g is the ground state.
The 5S+g multiplet is regular,
has no interpenetrating states,
and its components are evenly spaced.
The rest of the spectrum is complicated.
The first charge transfer states are the 3su1 du2fu2 3pu1 7P0+g
states.
That state for CmO2 2+ lies 16,000 cm-1 higher than for AmO2 +.
The parenthesized values on the left are assignments of the observed
AmO2 + transitions.

Figure 2:
AmO2 + MRSOCIS/cc-pVDZ Electronic Spectrum
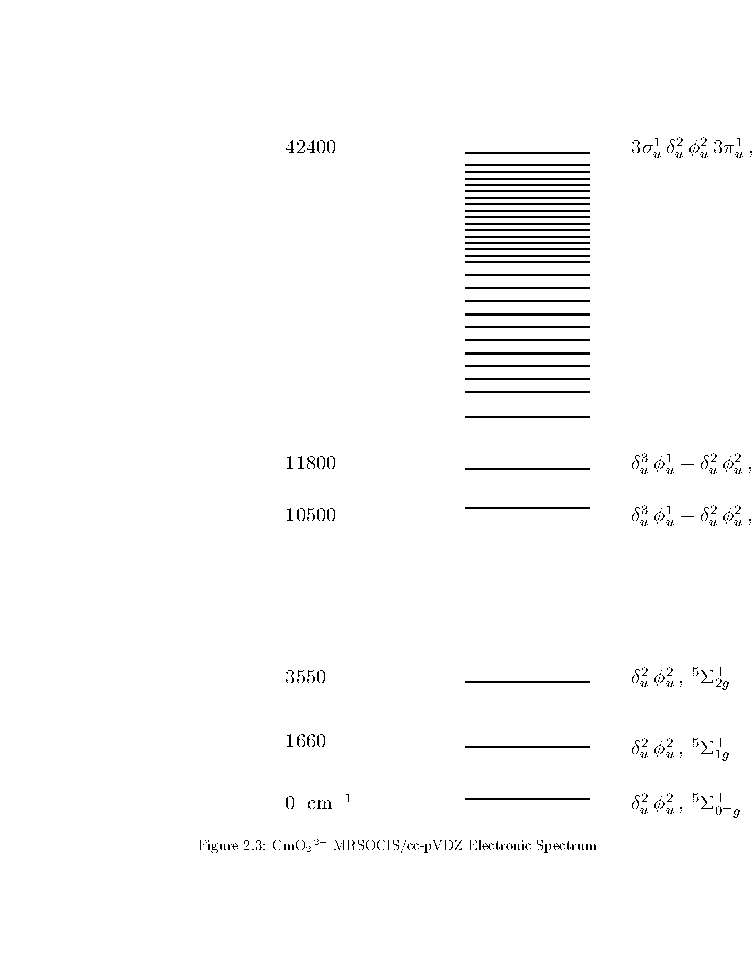
Figure 3:
CmO2 2+ MRSOCIS/cc-pVDZ Electronic Spectrum
At the SCF/cc-pVDZ level the du2fu2 5S+0+g
equilibrium bond distances are 1.55 Å
and 1.58 Å for AmO2 + and CmO2 2+.
At the MRSOCISD/cc-pVDZ level the du2fu2 5S+0+g
ground state
Re's are 1.59 and 1.62 Å for AmO2 + and CmO2 2+.
Symmetric stretch frequencies,
Table ,
are 932 and 1103 cm-1, respectively.
The 28% relative error for AmO2 + is large.
| | AnO2 2+ | AnO2 1+ |
| An | Exp. [] | Calc. | Exp. [,] | Calc. |
| U | 872 cm-1 | 1010 | - | - |
| Np | 863 | 1059 | 767 | 913 |
| Pu | 835 | 996 | 748 | - |
| Am | 796 | 1240 | 730 | 932 |
| Cm | - | 1103 | - | - |
Table 2:
Symmetric Stretch Frequencies
2.3
CmO2 +
MCSCF calculations yield a du2fu2 3pu1 6Pu ground state.
The MRSOCIS/cc-pVDZ electronic spectrum
is listed in Figure .
du2fu2 3pu1 6Pu is the ground state.
Its W value is probably 3/2;
L + S is probably either -3/2 or +3/2.
The rest of the spectrum is complicated.
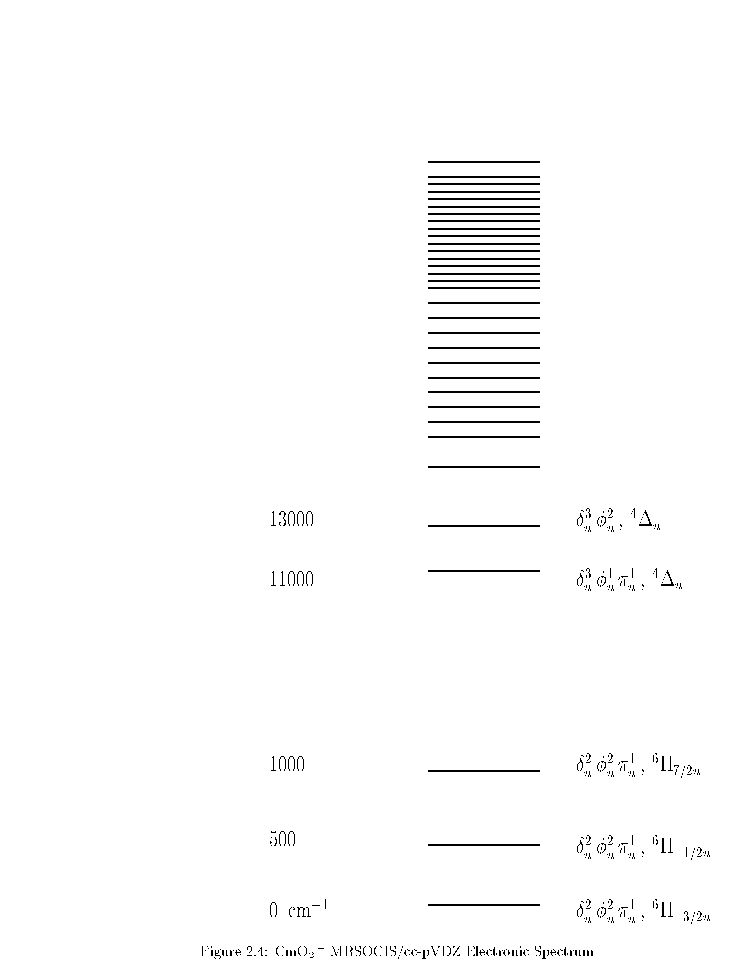
Figure 4:
CmO2 + MRSOCIS/cc-pVDZ Electronic Spectrum
3
Conclusions and Future Work
The ground state of the americyl cation is
du2fu1 4F3/2u.
The first charge transfer state is
3su1 du2fu2 6S5/2u.
The ground states of the isoelectronic dioxoactinide(V) and curyl
cations are du2fu2 5S+0+g.
The first charge transfer states are
3su1 du2fu2 3pu1 7P0+g.
Agreement with likely experimental charge transfer excitation
energies is good for those
that have been observed experimentally, namely, AmO2 2+ and AmO2 +.
Other electronic f ¬ f
transitions have been tentatively assigned.
Software development on computing transition moments,
in progress in this group, will help
in further study of these important spectral features.
Symmetric stretch frequencies, Table 2,
have been calculated for the ground states.
Obvious avenues of improvement of this work have been outlined in
Section .
Further study might include the dioxocurium(V) cation.
The AmO2 + and CmO2 2+ charge transfer states,
3su1 du2fu2 3pu1 7P0+g, suggest that CmO2 + may have a
du2fu2 3pu1 6P-3/2g ground state.
If that is the case then curiousity demands a study of
einsteinyl, EsO2 2+, to determine whether its ground state is an octet.
NEXT