Mail:
Dept. of Chemistry
Ohio State University
100 W. 18th Ave.
Columbus, OH 43210
Office:
412 CBEC
Email:
herbert@
chemistry.ohio-state.edu
|
|
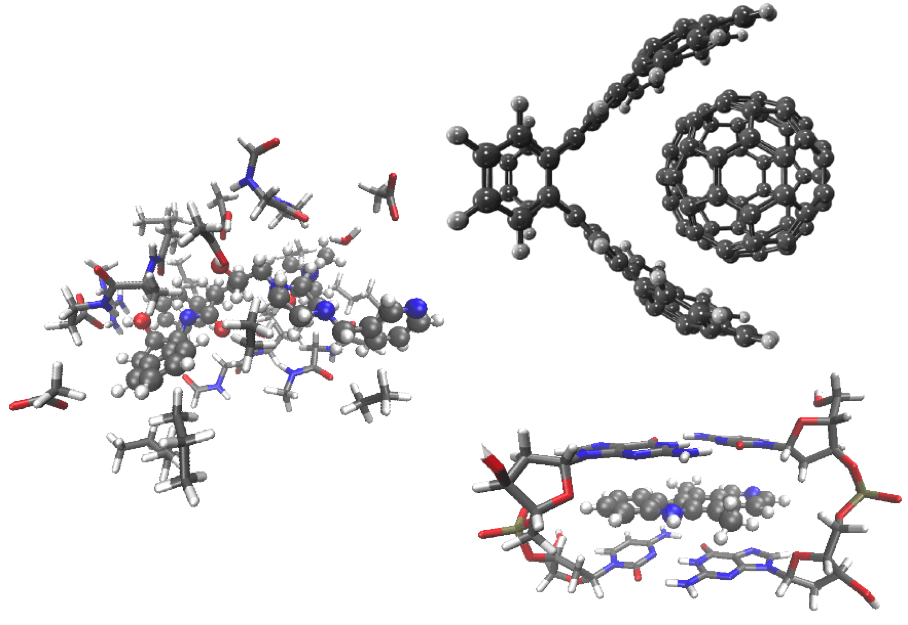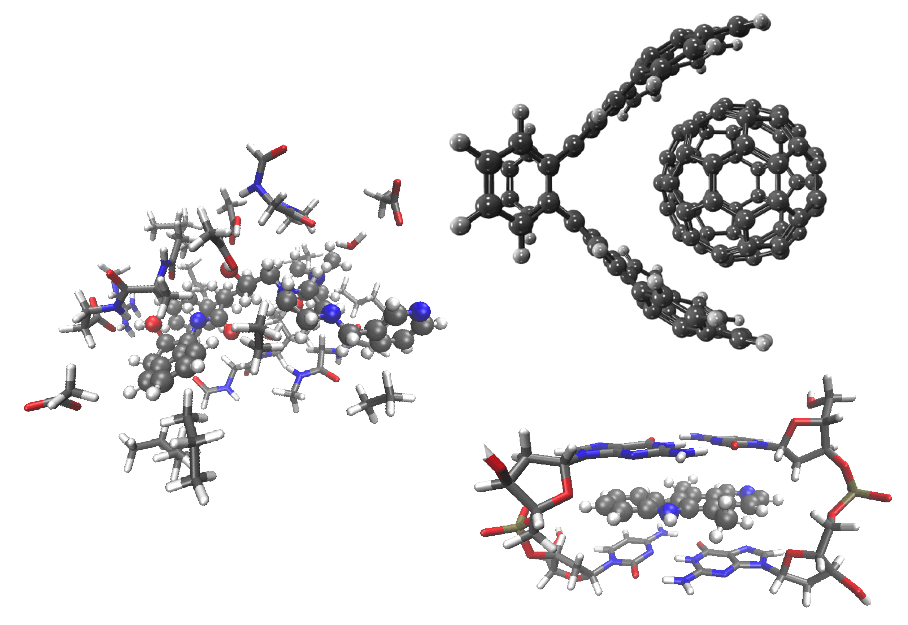
Supramolecular complexes (clockwise from top right):
C60-buckycatcher complex, DNA intercalation complex, and a drug molecule (ball-and-stick model) with nearby amino acids in an enzyme active site. |
Accurate and efficient calculation of intermolecular interactions is a challenging problem for electronic structure theory. Although the van der Waals interaction is one of the most fundamental concepts in chemistry, from a quantum-mechanical point of view it arises purely from electron correlation, and thus demands a high-level theoretical treatments if it is to be described properly. However, such calculations are seldom amenable to large collections of monomers, and certainly not to liquids, and thus cannot be used to describe the important cooperative (non-additive or many-body) effects present in clusters and in the condensed phase. We are working to develop ab initio methods for many-body systems that exploit monomer-based SCF calculations whose cost scales linearly with the number of monomers. These calculations are then coupled to two- and three-body treatments of the intermolecular interactions, to obtain accurate many-body interaction energies (comparable to the best supersystem ab initio calculations) at dramatically reduced cost. We have written short reviews of many-body methods for non-covalent interactions. and "extended" symmetry-adapted perturbation theory (XSAPT).
|
|
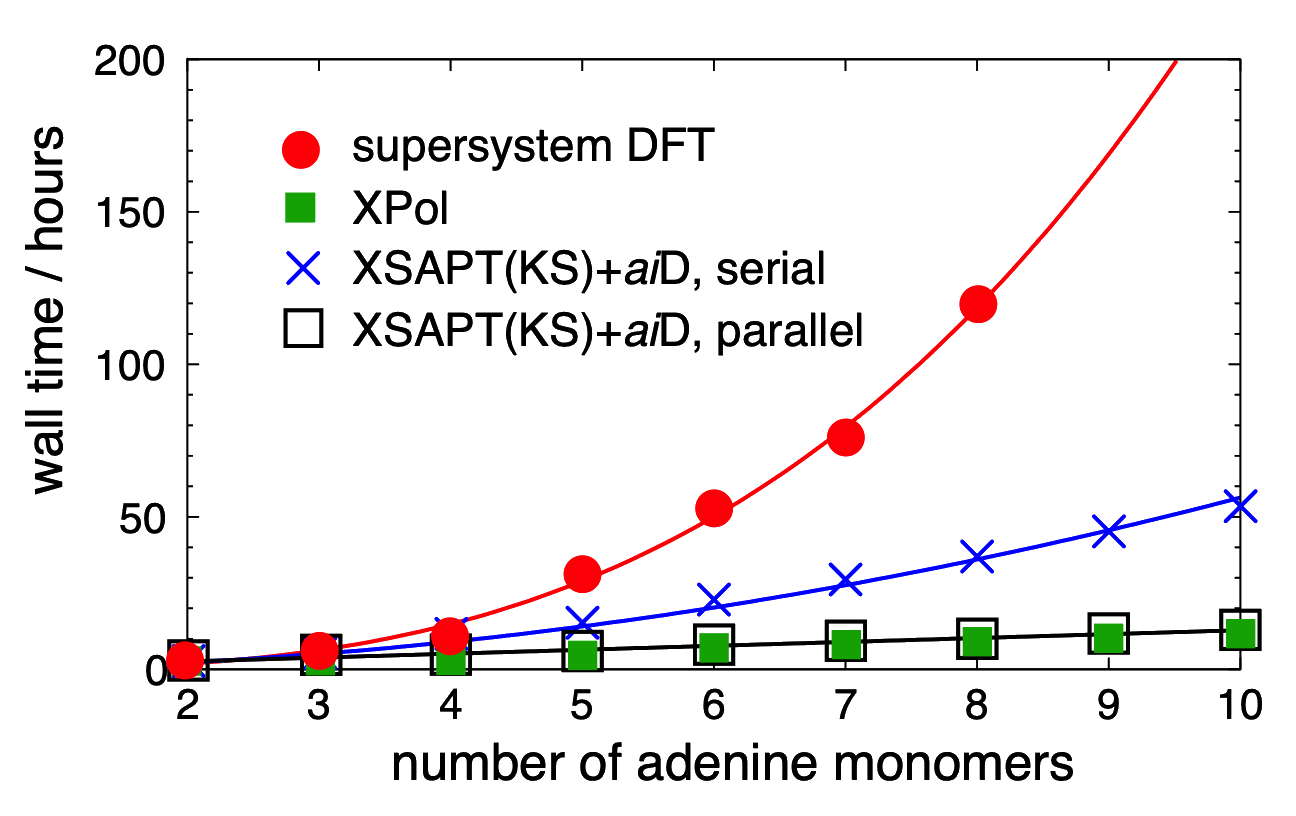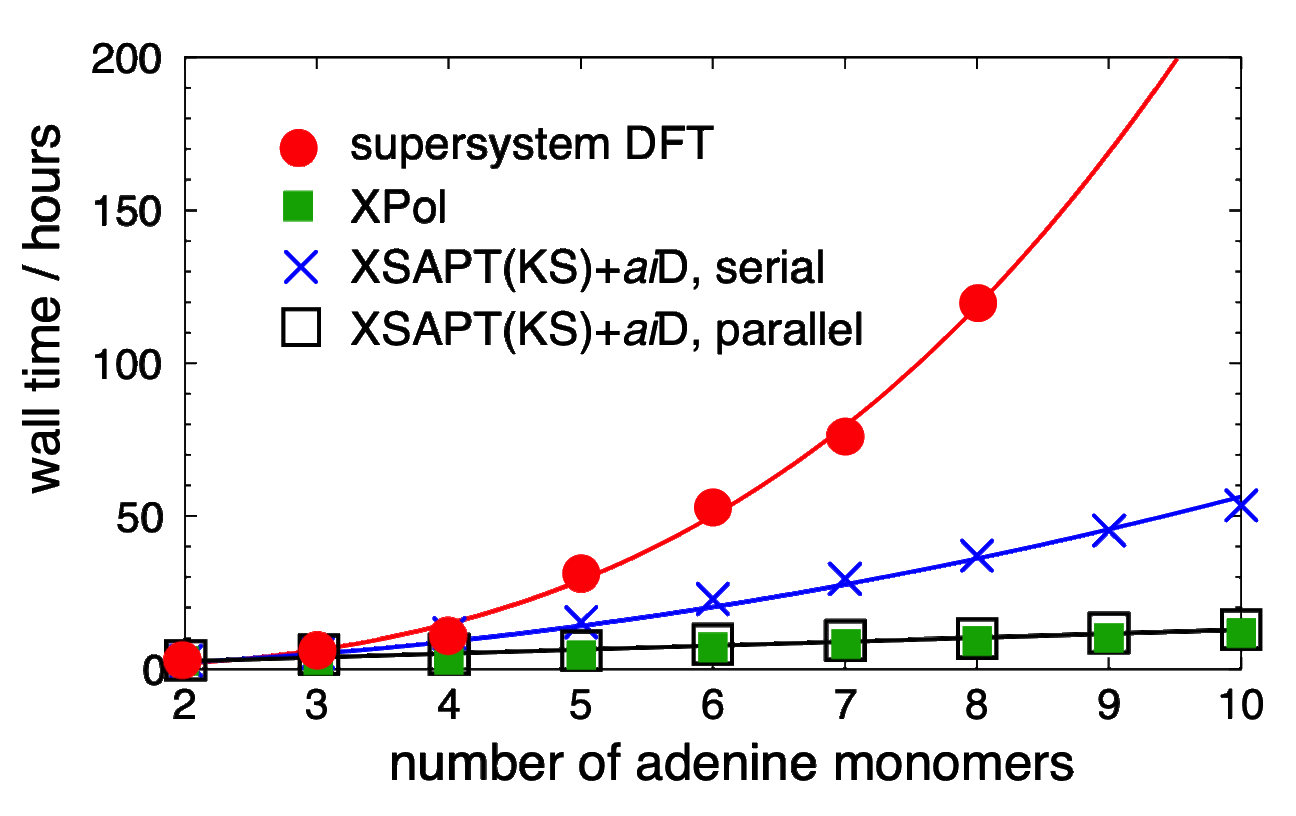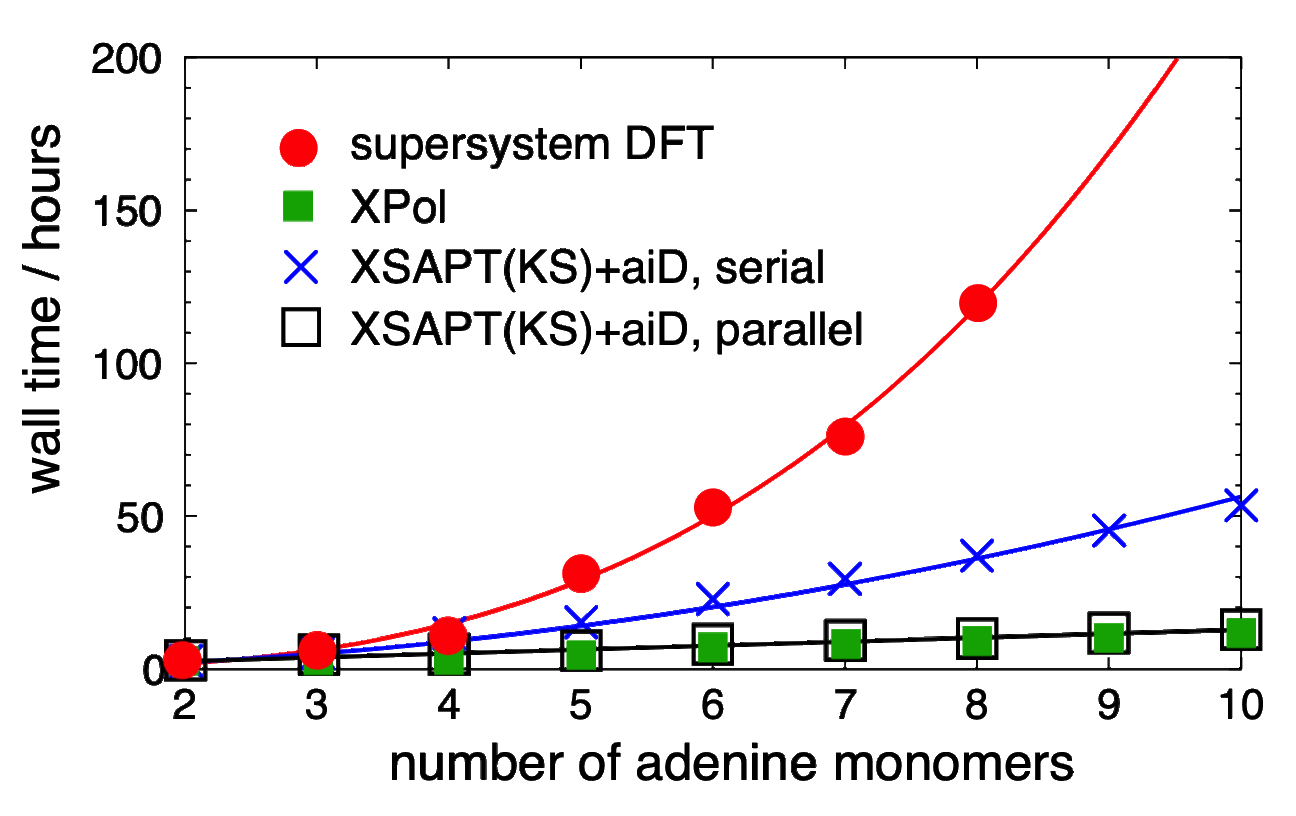
DFT+D and XSAPT(KS)+aiD timings for polyadenine |
We have recently introduced a family of ab initio methods for computing interaction energies in many-body systems. This approach is based on a self-consistent, monomer-based treatment of polarization (the "explicit polarization" or "XPol" procedure), which can be performed efficiently even in many-body systems, and which generates monomer wave functions that are polarized as appropriate for the many-body environment. These XPol monomer wave functions then form the basis states for a subsequent, pairwise version of symmetry-adapted perturbation theory (SAPT) to describe the remaining contributions to the intrmolecular interaction, primarily dispersion and exchange repulsion. The composite XSAPT method extends the well-known SAPT methodology to systems composed of more than two monomer units.
|
|
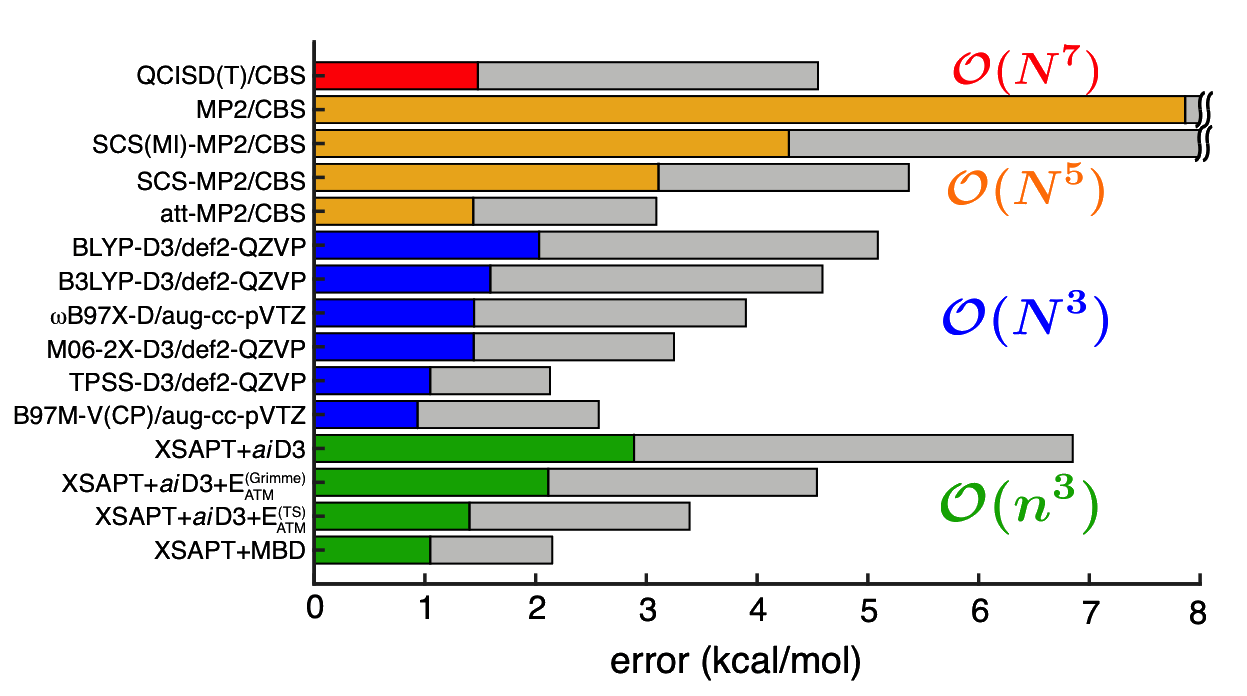
Error statistics for interaction energies in the L7 data set of dispersion-bound supramolecular complexes. (Colored bars are mean errors and gray bars are max errors.) |
Furthermore, replacing the SAPT dispersion terms with "ab initio dispersion potentials" ("+aiD"), which are classical potentials fit to ab initio dispersion data. We use Kohn-Sham DFT as a low-cost means to treat intramolecular electron correlation. The resulting method, which we call XSAPT(KS)+aiD, exhibits cubic-scaling cost, both with respect to the number of monomer units as well as with respect to the size of those monomer units. Moreover, by exploiting the embarrassingly parallel nature of the XSAPT approach, the total wall time for XSAPT(KS)+aiD calculations can be made to scale linearly with respect to the number of monomer units. Despite this favorable scaling, the accuracy of XSAPT(KS)+aiD is better than 0.5 kcal/mol for the S22 and S66 databases of benchmark [CCSD(T)/CBS] non-covalent binding energies. This level of accuracy, which we achieve using triple-ζ basis sets, is far better than MP2-based methods for dispersion-dominated complexes, even while the cost of MP2 scales as O(N5) with respect to the size of the total (super)system. In contrast, the cost of XSAPT calculations grows only as O(n3) with respect to the size of the largest monomer. As a result, XSAPT is even faster than supramolecular DFT+D approaches, without the need for counterpoise correction.
An even newer approach, XSAPT+MBD, replaces the +aiD dispersion potentials with something closer to a truly first-principles approach based on using atoms-in-molecules polarizabilties to compute C6 and C8 coefficients that include many-body effects. XSAPT+MBD achieves an accuracy of ~1 kcal/mol with respect to CCSD(T)/CBS benchmarks, and demonstrates that many-body dispersion effects are always important in large systems, even those that lack "canonical" dispersion interactions such as π-stacking.

|

|