Mail:
Dept. of Chemistry
Ohio State University
100 W. 18th Ave.
Columbus, OH 43210
Office:
412 CBEC
Email:
herbert@
chemistry.ohio-state.edu
|
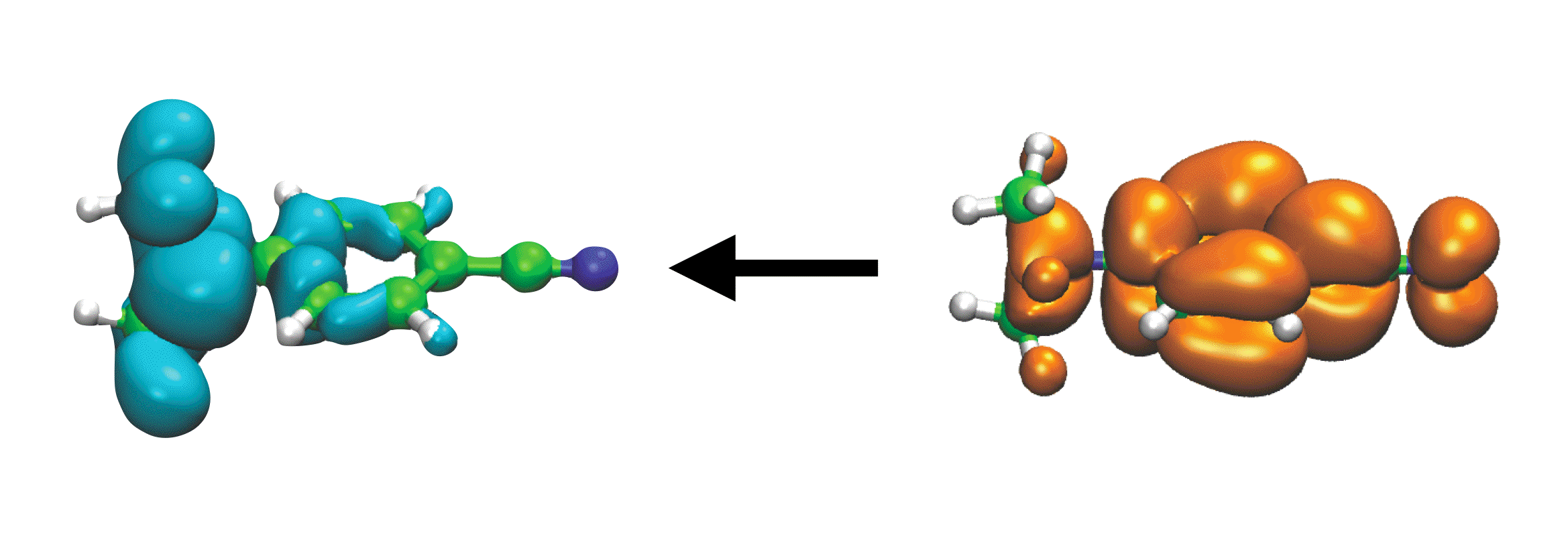
|
S0 ← S1 emission in the DMABN molecule, which exhibits intramolecular CT character |
Electronic excitation is characterized by an instantaneous (and possibly drastic) change in the electron density of the molecular or functional group that absorbs the photon. In particular, intramolecular charge-transfer excited states such as those present in "donor-π-acceptor" or "push-pull" chromophores, can change the dipole moment of the chromophore by 10 debye or more. In a condensed-phase environment, a proper description of such an event should allow the electronic degrees of freedom of the environment to remain in equilibrium with those of the chromophore, i.e., the chromophore and its environment should polarize one another in a self-consistent fashion. In addition, the most accurate molecular mechanics (MM) force fields are often polarizable ones. In the context of QM/MM calculations, the increased cost associated with using a polarizable force field should be insignificant, provided that the QM/MM algorithm is designed in a reasonable way. We are developing computationally-efficient QM/MM models that utilize polarizable MM force fields ("QM/polMM" models), in an effort to describe excited states in solution. We aim for solvation models that are accurate across the entire range of system sizes, from small clusters to bulk solvation. These new methods enable us to explore the role of solvent polarization, which is especially important for the description of charge-separated excited states, and to make contact with both condensed-phase electronic spectroscopy, as well as gas-phase cluster experiments. We have also redesigned periodic boundary conditions for QM/MM calculations, in order to deal with periodic images of the QM wavefunction in a robust way, even for large basis sets.
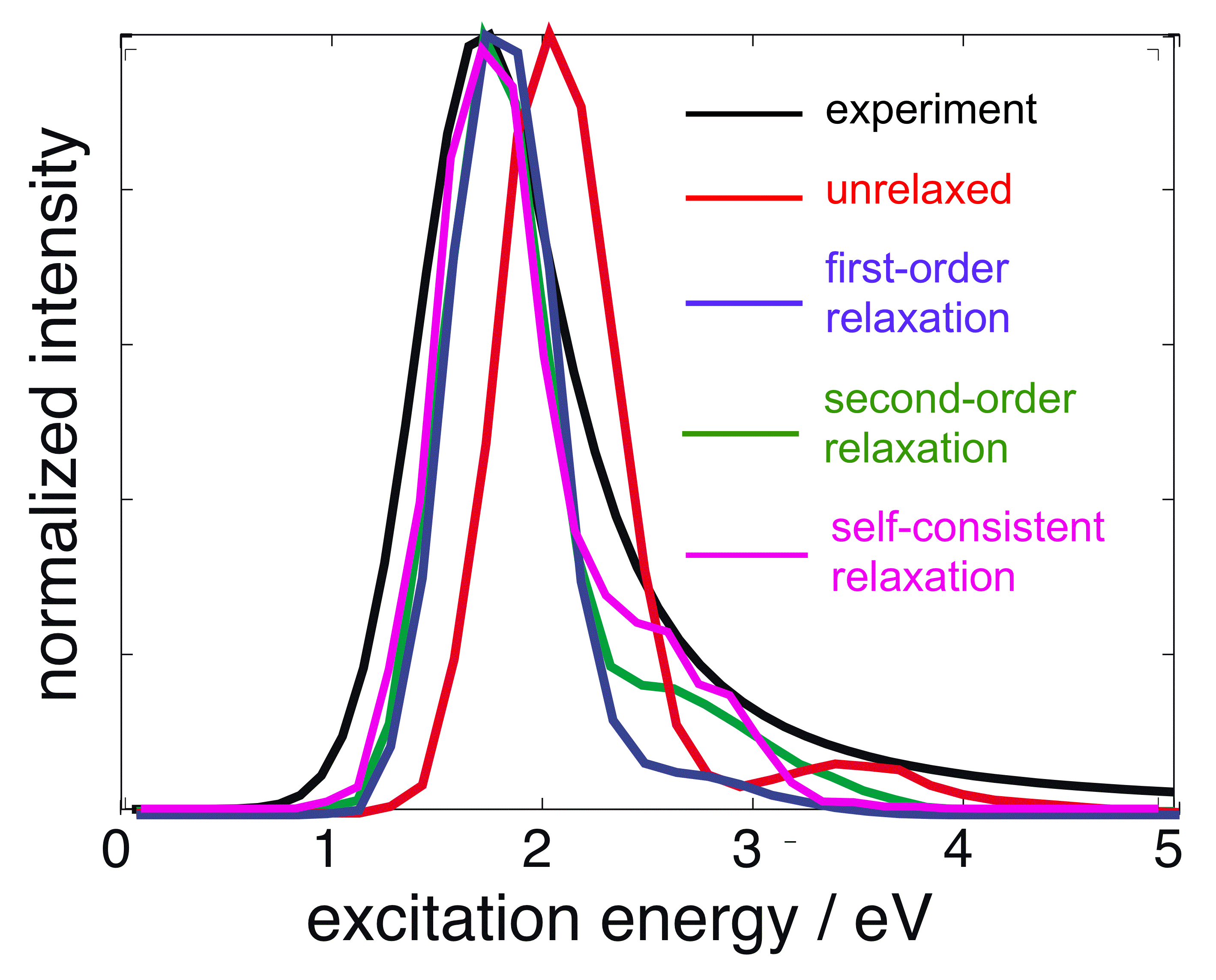
|
|
QM/polMM optical absorption spectra for the aqueous electron. Various approximations for excited-state relaxation of the solvent polarization are compared. |
We have recently developed a QM/polMM model for the aqueous electron, based on a real-space QM description of a single electron, in conjunction with the AMOEBA polarizable water model. Despite 25 years of detailed, atomistic simulations of this systems, ours is the first model that results in a significant "blue tail" in the optical absorption spectrum, in agreement with experiment. We find that this tail results from intensity-borrowing on the part of quasi-continuum states that are bound only by solvent polarization in the excited state. Such a mechanism is absent in any non-polarizable QM/MM model.
Owing to the complexity of calculating excited-state polarization in a self-consistent manner, we have developed perturbative approximations for the change in solvent polarization upon excitation of the wavefunction. Interestingly, we find that second-order perturbation theory (i.e., a correction to the wavefunction itself, not just the excitation energies) is needed in order to obtain qualitatively correct oscillator strengths. This further underscores the importance of treating the polarization of the environment in a realistic way.
Our calculations of the vertical electron binding energy for the aqueous electron, using the aforementioned QM/polMM model, are the first (and, to date, only) theoretical values that lie within the range of binding energies measured in recent liquid microjet experiments. Here, too, a self-consistent treatment of electron ↔ water polarization is crucial in order to obtain results that are in quantitative or semi-quantitative agreement with experiment. Our model predicts that solvent re-polarization upon vertical detachment of the excess electron reduces the binding energy by ≈ 1.4 eV, or about 40% of its value.