Mail:
Dept. of Chemistry
Ohio State University
100 W. 18th Ave.
Columbus, OH 43210
Office:
412 CBEC
Email:
herbert@
chemistry.ohio-state.edu
|
|
The s → p excitation of the aqueous electron, which is responsible for its optical absorption spectrum. |
Just as we are working to extend excited-state quantum chemistry into condensed-phase environments, we also aim to achieve an understanding of open-shell species (in both ground and electronic states) in liquid solution. Modern ultrafast spectroscopy provides a variety of new experimental tools to study radicals and ions in liquid solution, and we seek to develop comparable theoretical tools to understand observable spectroscopic signatures. Often for open-shell species, there are gaps in our knowledge of structural and dynamical aspects of solvation such as diffusion mechanisms, coordination numbers, or the extent to which solvent molecular orbitals contribute to the electronic structure of the radical in question. In liquid water, a detailed understanding of the spectroscopy of H atom, OH radical, and the "hydrated electron"—the three most important radicals generated when water is exposed to ionizing radiation—is necessary in order to provide a detailed picture of aqueous radiation chemistry. Further complicating the issue is the fact that radical reactivity can be very different at interfaces than in bulk solution. A complete, molecular-level understanding of these phenomena will require not only accurate quantum mechanics (since molecular mechanics force fields are often unavailable or unreliable for open-shell species) but also good statistical mechanics, i.e. proper averaging over bulk solvation environments, rather than the sort of "microsolvation" approaches that are commonly used in quantum-chemical calculations.
|
|
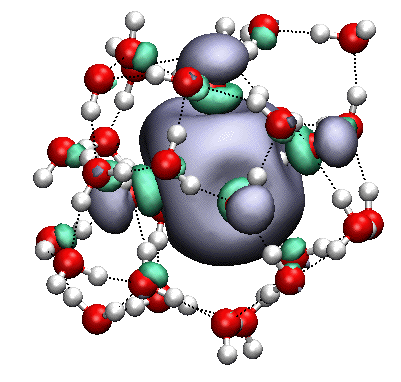
The singly-occupied MO of the anionic water cluster (H2O)31– |
The hydrated (or aqueous) electron, in particular, is both an important intermediate in radiation chemistry, but also a fascinating species in its own right, and one that our group has spent a great deal of effort trying to understand. The solute, e–, is a charged species and thereby disrupts the local hydrogen-bonding environment of liquid water (into the second solvation shell, according to our calculations), an effect that may not be properly capture by microsolvation approaches. At the same time, however, the ion is a fundamentally quantum-mechanical species, with a diffuse, polarizable wavefunction that is greatly influenced by the local solvation environment, so that accurate quantum mechanics calculations are required, along with a consistent treatment of water ↔ water and electron ↔ water polarization. A review of the importance of polarization can be found here.
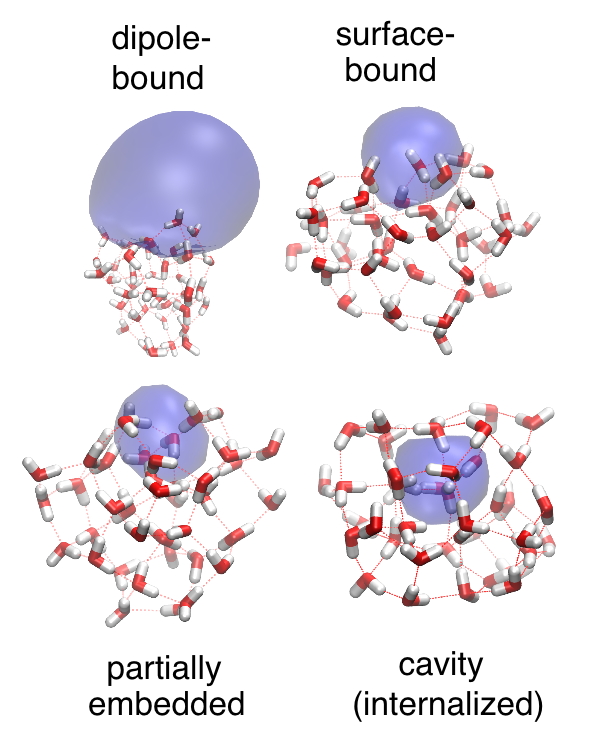
A detailed understanding of ion and radical solvation is also one of the principal aims of the community of physical chemists who study gas-phase clusters, and we have worked closely with experimental groups in this field, especially in the context of anionic water clusters, (H2O)n–. These have been studied in the gas phase for since the 1980s, and a wealth of experimental data are available, yet the interpretation of many of these data remains controversial. A hotly-debated question, for example, is the cluster size at which the transition from a surface-bound to a cavity-bound electron becomes thermodynamically spontaneous; we have put forward an assignment of experimental photoelectron spectra for (H2O)n– clusters that attempts to address this issue.
|
|
Binding motifs in hydrated- electron clusters |
Our work on the hydrated electron was initially motivated by the experimental studies of the vibrational and photo-electron spectroscopy of anionic water clusters. Using a new one-electron model developed recently by our group, which is accurate enough to predict electron detachment energies within ≈0.1 eV but affordable enough to perform simulations, we have recently proposed assignments for the various isomer series that have been observed in photo-electron spectroscopy experiments. The identify of these various isomers—in particular, which one (if any) represents a "cavity-bound" isomer analogous to the bulk aqueous electron—has been a controversial topic for over two decades.
|
|
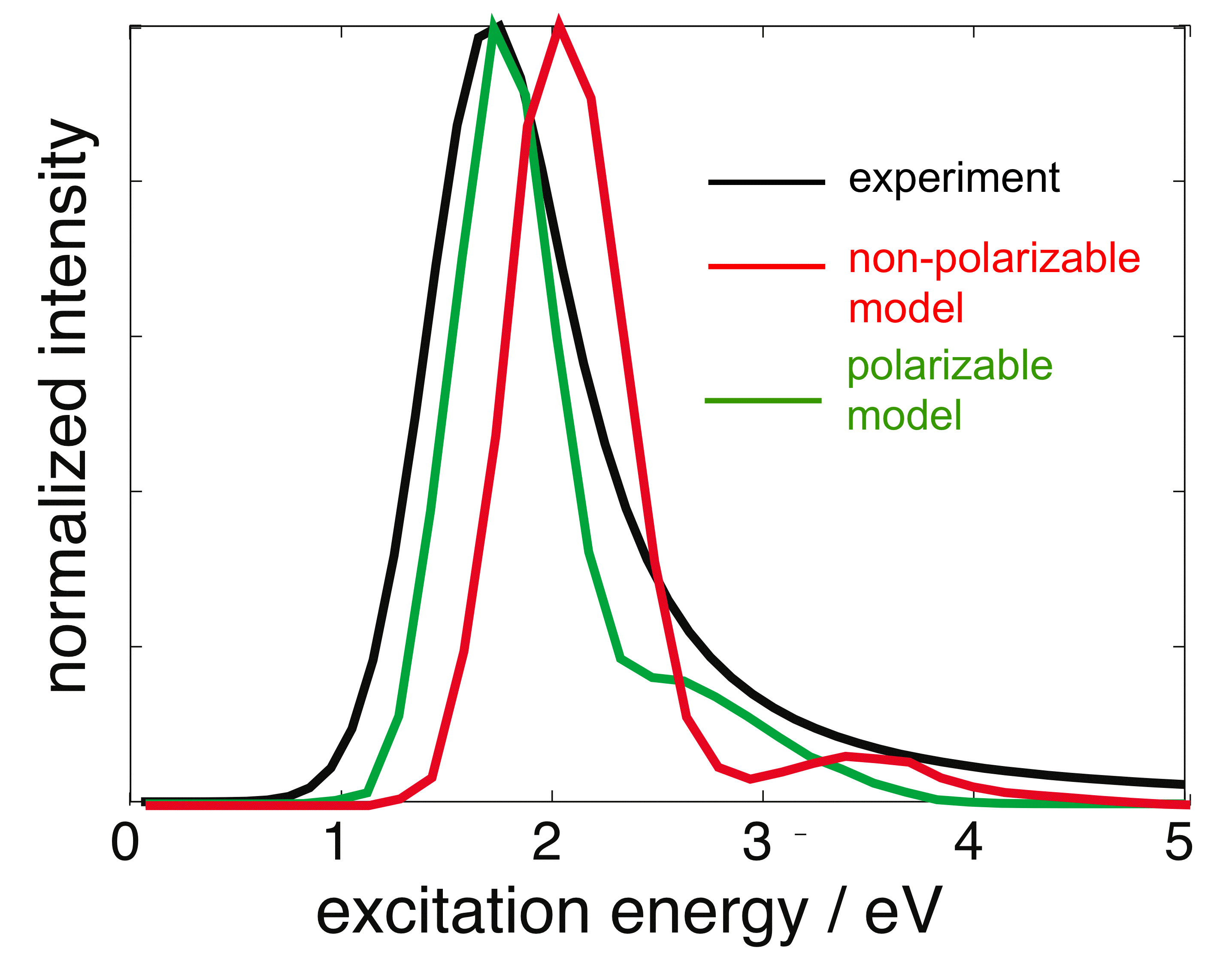
Optical absorption spectra for the aqueous electron, calculated using two different QM/MM models. |
Our one-electron model entails an essentially exact numerical solution of a one-electron Schrödinger equation (on a real-space grid), coupled to a polarizable water force field. This represents a special case of more general polarizable QM/MM methods developed in our group, and in the context of the hydrated electron, it represents the first attempt to treat QM/MM polarization in a fully self-consistent manner. In fact, despite 25 years of detailed, atomistic simulations of the aqueous electron, our model is the first to predict and explain the "blue tail" that is observed, experimentally, in the optical absorption spectrum. We find that this tail results from intensity-borrowing by quasi-continuum states that are bound only by solvent polarization in the excited state, a mechanism that is necessarily absent in any non-polarizable QM/MM model. Our calculations of the vertical electron binding energy for the aqueous electron are also the first (and, to date, only) theoretical values that lie within the range of binding energies measured in recent liquid microjet experiments. Here, too, a self-consistent treatment of electron ↔ water polarization is crucial in order to obtain results that are in quantitative or semi-quantitative agreement with experiment. Our model predicts that solvent re-polarization upon vertical detachment of the excess electron reduces the binding energy by ≈ 1.4 eV, or about 40% of its value.
Recently, the long-held view that the aqueous electron carves out an occupies a void in the structure of bulk liquid water has been questioned, on the basis of simulations employing an alternative one-electron model. We have recently addressed this issue using a combination of one-electron simulations and time-dependent density functional theory calculations, using long-range-corrected density functionals developed in our group, and we implemented resonance Raman capabilities in order to make contact with experiments. At present, we feel that the weight of the data support the cavity-centric viewpoint, as we reported in a "Perspective" that provides an overview of both experiments and simulations.