Mail:
Dept. of Chemistry
Ohio State University
100 W. 18th Ave.
Columbus, OH 43210
Office:
412 CBEC
Email:
herbert@
chemistry.ohio-state.edu
|
|
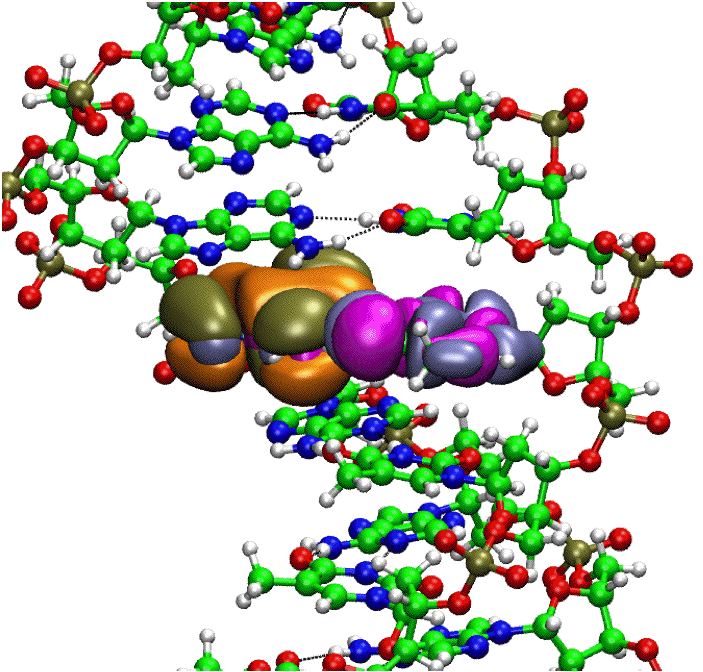
The HOMO and LUMO of a base pair in DNA. |
The excited electronic states of many small chromophores are well-characterized in the gas phase, but much less is known regarding how these states are perturbed by a solvent, or by some other condensed-phase environment such as the interior of a protein. The location of conical intersections, as well as energy barriers along an excited-state reaction pathway, may be profoundly different in the condensed phase than they are in the gas phase, and these details ultimately dictate whether the energy deposited into a chromophore by a visible or UV photon manifests as fluorescence, radiationless decay to the ground state (internal conversion), or else initiates some excited-state (photochemical) reaction. To understand condensed-phase electronic spectroscopy at a theoretical level, and to simulate condensed-phase photochemistry and photophysics, it is crucial that we develop methods that afford realistic models of the environment of a chromophore, including possible electronic coupling to other chromophores. Our group is working to develop and refine ab initio simulation methods that can be applied to excited-state reactions in the condensed phase.
Although the methods that we seek to develop are general, a primary motivation has been to understand the nature of energy flow following UV excitation in DNA. An detailed mechanistic understanding of DNA photochemistry is important insofar as photochemical isomerization and strand cleavage in this molecule are potentially mutagenic. At the same time, ultrafast spectroscopy has recently revealed a complex set of time scales for DNA photophysics, which at present are not completely understood. Of particular interest is the role of excitonic coupling between the DNA nucleobases (each of which is a UV chromophore), which can lead to excited-state wavefunctions that are delocalized over two or more nucleobases. As such, the full complexity of the electronic wavefunctions is only realized in systems that are quite large, by the standards of contemporary quantum chemistry. In addition, the role of charge-transfer excited states (in which an electron is transferred, e.g., from the HOMO of one nucleobase to the LUMO of its hydrogen-bonded or π-stacked partner) has been hotly debated, as such states are absent in the gas phase but may be stabilized in polar solvents.
|
|
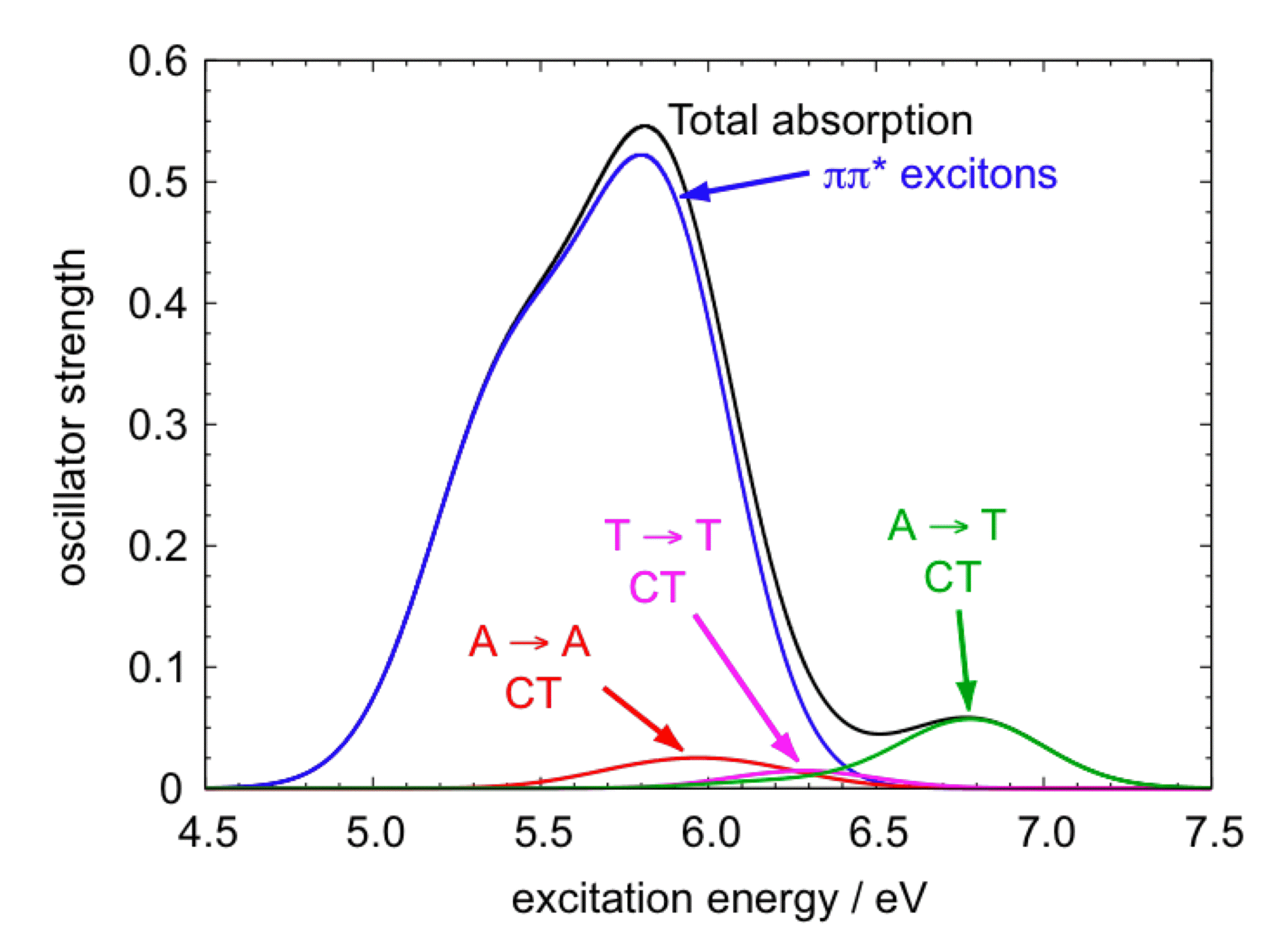
Calculated absorption spectrum of duplex Ade2:Thy2 in aqueous solution decomposed into excitonic bands and CT bands. |
Our efforts to understand the excited states of DNA, based on quantum chemistry calculations, draw upon a wide variety of computational tools, many of them developed in our group over the last few years. Primarily, these include the parameterization of certain "long-range-corrected" density functionals that, in the context of time-dependent density function theory (TD-DFT), can provide a balanced treatment of both localized valence excited states (e.g., nπ*, ππ*, or πσ* states), as well as charge-transfer (CT) states. Description of the latter has long been problemtic within TD-DFT, and we have developed what was arguably the first functional to predict both valence and CT excitation energies with comparable accuracy. A balanced treatment is crucial, if we are to shed light on the existence of CT excited states in DNA, and our work provides the first compelling computational evidence that optically-dark CT excited states do exist in the low-energy region of DNA's UV spectrum. These states exist alongside the "excitonic" bright states that are formed from linear combinations of localized ππ* excitations on the individual nucleobases (e.g., HOMO → LUMO or HOMO-1 → LUMO excitations). In the future, we expect that our group's recent developments in the areas of continuum solvation models and polarizable QM/MM models can be used, in the context of TD-DFT calculations, to provide a detailed picture of excited state potential surfaces in DNA oligomers.
|
|
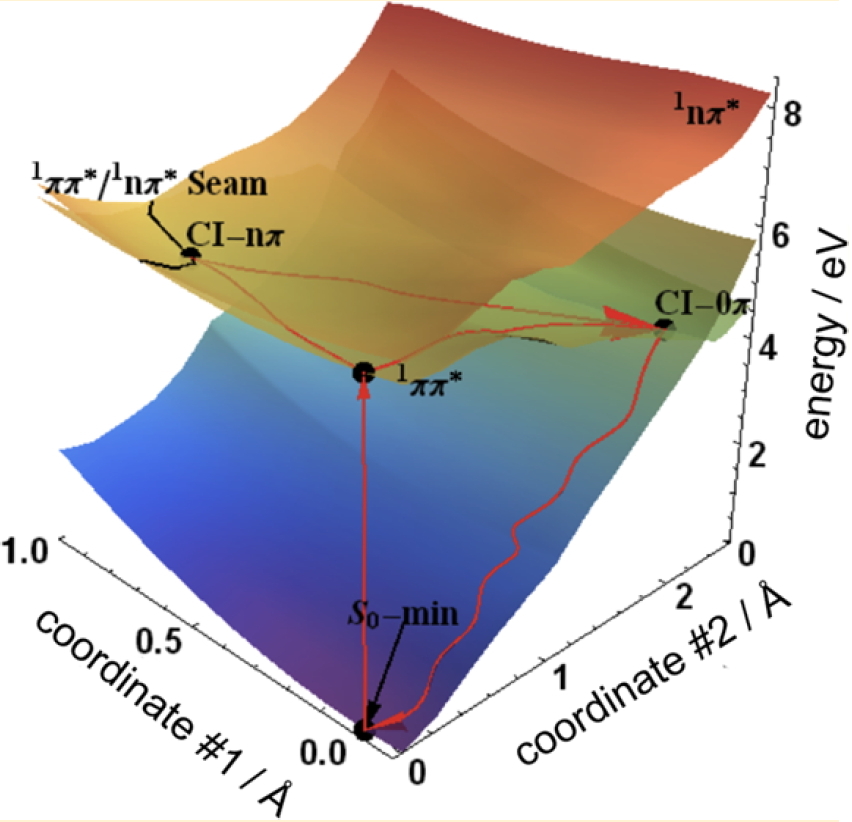
Potential energy surfaces for aqueous uracil, indicating the photophysical relaxation pathway. |
|
|
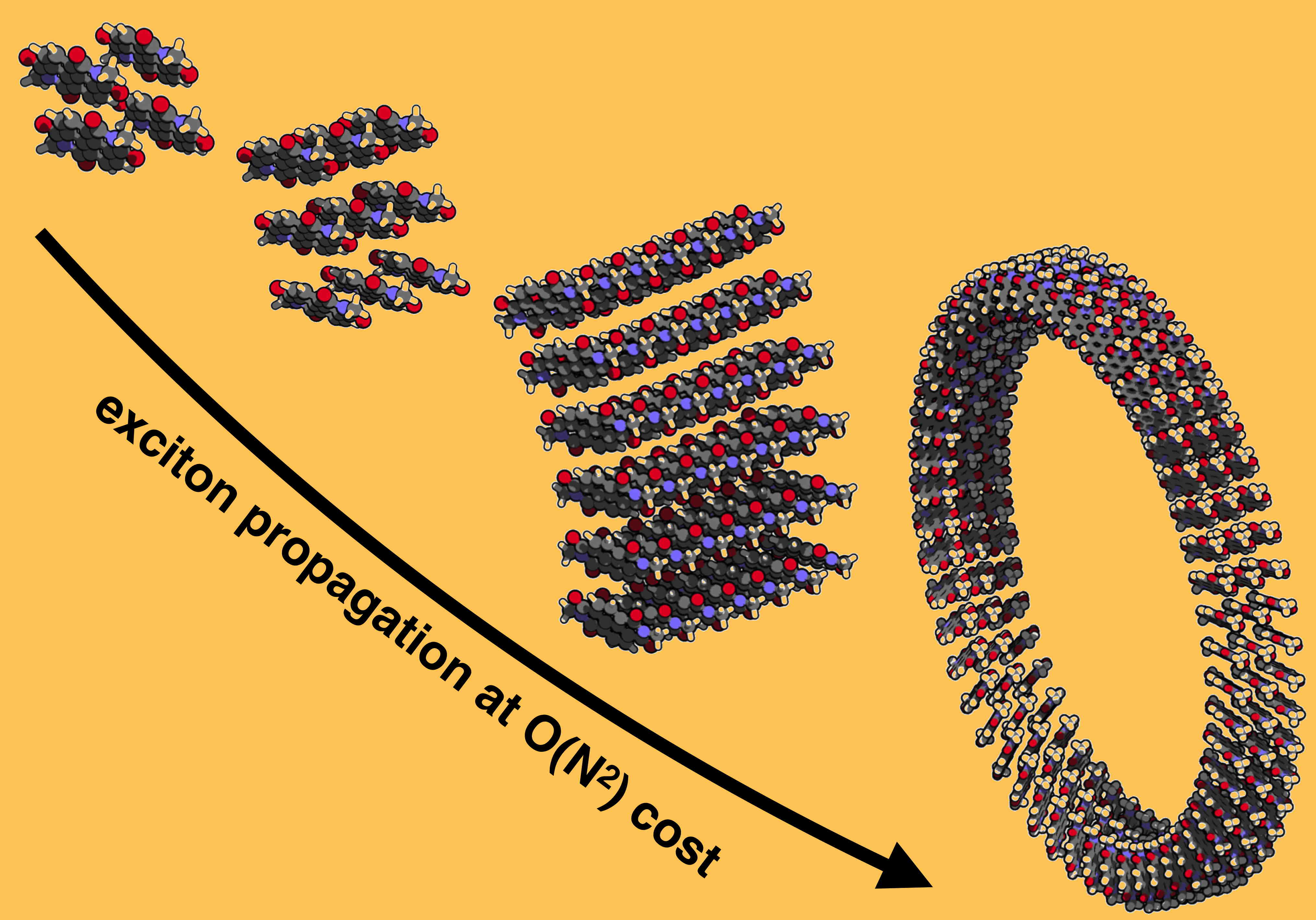
Models of a self-assembled organic semi-conductor nanotube. |